Recommendations for UK Biobank analysis
regenie is ideally suited for large-scale analyses such as 500K UK Biobank (UKBB) data, where records are available for thousands of phenotypes.
We provide below a few guidelines on how to perform such analysis on the UKBB files that all UKBB approved researchers have access to.
Pre-processing
We will first go over important steps to consider before running regenie.
Selection of traits
regenie can perform whole genome regression on multiple traits at once, which is where higher computational gains are obtained.
As different traits can have distinct missing patterns, regenie uses an imputation scheme to handle missing data. From the real data applications we have performed so far with traits having up to ~20% (for quantitative) and ~5% (for binary) missing observations, our imputation scheme resulted in nearly identical results as from discarding missing observations when analyzing each trait separately (see the paper for details). Hence, we recommend to analyze traits in groups that have similar missingness patterns with resonably low amount of missingness (<15%).
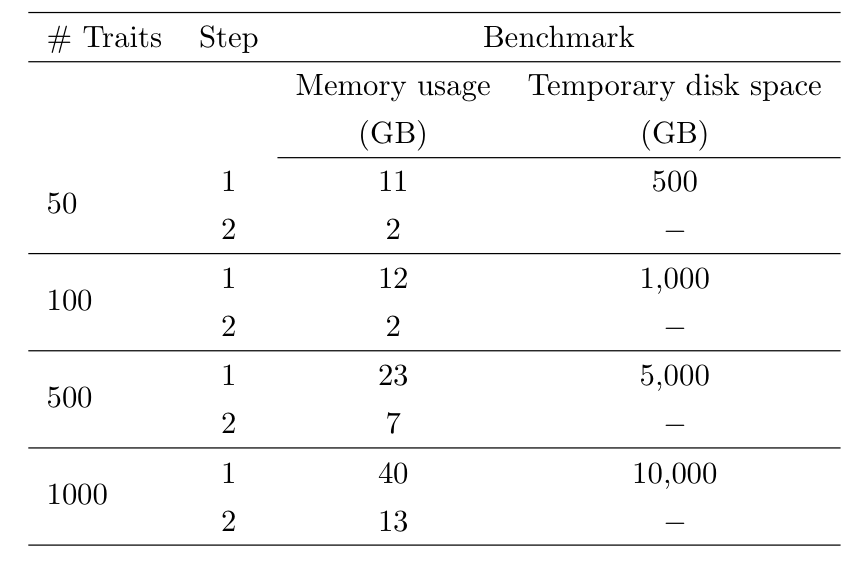
The number of phenotypes in a group will affect the computational resources required and the table below shows typical computational requirements based on using 500,000 markers in step 1 split in blocks of 1000 and using blocks of size 200 when testing SNPs in step 2. The estimates are shown when step 1 of regenie is run in low-memory mode so that within-block predictions are temporarily stored on disk (see Documentation).
In the following sections, we'll assume traits (let's say binary) and covariates
used in the analysis have been chosen and data are in files
ukb_phenotypes_BT.txt and ukb_covariates.txt,
which follow the format requirement for regenie (see Documentation).
Preparing genotype file
Step 1 of a regenie run requires a single genotype file as input; we recommend using array genotypes for this step. The UKBB genotype files are split by chromosome, so we recommend using PLINK to merge the files using the following code.
NOTE: please change XXX to you own UKBB application ID number
rm -f list_beds.txt
for chr in {2..22}; do echo "ukb_cal_chr${chr}_v2.bed ukb_snp_chr${chr}_v2.bim ukbXXX_int_chr1_v2_s488373.fam" >> list_beds.txt; done
plink \
--bed ukb_cal_chr1_v2.bed \
--bim ukb_snp_chr1_v2.bim \
--fam ukbXXX_int_chr1_v2_s488373.fam \
--merge-list list_beds.txt \
--make-bed --out ukb_cal_allChrs
Exclusion files
Quality control (QC) filters can be applied using PLINK2 to filter out samples and markers in the genotype file prior to step 1 of regenie.
Note: regenie will throw an error if a low-variance SNP is included in the step 1 run. Hence, the user should run adequate QC filtering prior to running regenie to identify and remove such SNPs.
For example, to filter out SNPs with minor allele frequency (MAF) below 1%, minor allele count (MAC) below 100, genotype missingess above 10% and Hardy-Weinberg equilibrium p-value exceeding , and samples with more than 10% missingness,
plink2 \
--bfile ukb_cal_allChrs \
--maf 0.01 --mac 100 --geno 0.1 --hwe 1e-15 \
--mind 0.1 \
--write-snplist --write-samples --no-id-header \
--out qc_pass
Step 1
We recommend to run regenie using multi-threading (8+ threads) which will
decrease the overall runtime of the program.
As this step can be quite memory intensive (due to storing block predictions),
we recommend to use option --lowmem, where the number of phenotypes analyzed
will determine how much disk space is required (see table above).
Running step 1 of regenie (by default, all available threads are used)
./regenie \
--step 1 \
--bed ukb_cal_allChrs \
--extract qc_pass.snplist \
--keep qc_pass.id \
--phenoFile ukb_phenotypes_BT.txt \
--covarFile ukb_covariates.txt \
--bt \
--bsize 1000 \
--lowmem \
--lowmem-prefix tmpdir/regenie_tmp_preds \
--out ukb_step1_BT
For P phenotypes analyzed, this will generate a set of $P$ files ending with .loco
which contain the genetic predictions using a LOCO scheme that will be needed for step 2,
as well as a prediction list file ukb_step1_BT_pred.list, which lists
the names of these predictions files and can be used as input for step 2.
Step 2
As step 1 and 2 are completely decoupled in regenie, you could either use all the traits for testing in step 2 or select a subset of the traits to perform association testing. Furthermore, you can use the same Step 1 output to test on array, exome or imputed variants; below, we will illustrate testing on imputed variants.
Step 2 of regenie can be run in parallel across chromosomes so if you have access to multiple machines, we recommend to split the runs over chromosomes (using 8+ threads).
Running regenie tesing on a single chromosome (here chromosome 1) and using the fast Firth correction as fallback for p-values below 0.01
./regenie \
--step 2 \
--bgen ukb_imp_chr1_v3.bgen \
--ref-first \
--sample ukbXXX_imp_chr1_v3_s487395.sample \
--phenoFile ukb_phenotypes_BT.txt \
--covarFile ukb_covariates.txt \
--bt \
--firth --approx --pThresh 0.01 \
--pred ukb_step1_BT_pred.list \
--bsize 400 \
--split \
--out ukb_step2_BT_chr1
This will create separate association results files for each phenotype as ukb_step2_BT_chr1_*.regenie.
When running the SKAT/ACAT gene-based tests, we recommend to use at most 2 threads and instead parallelize the runs over partitions of the genome (e.g. groups of genes).