Getting started
To run regenie, use the command ./regenie on the command line,
followed by options and flags as needed.
To get a full list of options use
./regenie --help
The directory examples/ contains some small example files that are
useful when getting started. A test run on a set of binary traits can be achieved by the
following 2 commands.
In Step 1, the whole genome regression model is fit to the traits, and a set of genomic predictions are produced as output
./regenie \
--step 1 \
--bed example/example \
--exclude example/snplist_rm.txt \
--covarFile example/covariates.txt \
--phenoFile example/phenotype_bin.txt \
--remove example/fid_iid_to_remove.txt \
--bsize 100 \
--bt --lowmem \
--lowmem-prefix tmp_rg \
--out fit_bin_out
In Step 2, a set of imputed SNPs are tested for association using a Firth logistic regression model
./regenie \
--step 2 \
--bgen example/example.bgen \
--covarFile example/covariates.txt \
--phenoFile example/phenotype_bin.txt \
--remove example/fid_iid_to_remove.txt \
--bsize 200 \
--bt \
--firth --approx \
--pThresh 0.01 \
--pred fit_bin_out_pred.list \
--out test_bin_out_firth
One of the output files from these two commands is included in example/test_bin_out_firth_Y1.regenie.
Basic options
Input
| Option | Argument | Type | Description |
|---|---|---|---|
--bgen, --bed, --pgen |
FILE | Required | Input genetic data file. Either BGEN file eg. file.bgen, or bed/bim/fam prefix that assumesfile.bed, file.bim, file.fam exist, or pgen/pvar/psam prefix that assumesfile.pgen, file.pvar, file.psam exist |
--sample |
FILE | Optional | Sample file corresponding to input BGEN file |
--bgi |
FILE | Optional | Index bgi file corresponding to input BGEN file |
--ref-first |
FLAG | Optional | Specify to use the first allele as the reference allele for BGEN or PLINK bed/bim/fam file input [default is to use the last allele as the reference] |
--keep |
FILE | Optional | Inclusion file that lists individuals to retain in the analysis |
--remove |
FILE | Optional | Exclusion file that lists individuals to remove from the analysis |
--extract |
FILE | Optional | Inclusion file that lists IDs of variants to keep |
--exclude |
FILE | Optional | Exclusion file that lists IDs of variants to remove |
--extract-or |
FILE | Optional | Inclusion file that lists IDs of variants to keep regardless of minimum MAC filter |
--exclude-or |
FILE | Optional | Exclusion file that lists IDs of variants to remove unless MAC is above threshold |
--phenoFile |
FILE | Required | Phenotypes file |
--phenoCol |
STRING | Optional | Use for each phenotype you want to include in the analysis |
--phenoColList |
STRING | Optional | Comma separated list of phenotypes to include in the analysis |
--eventColList |
STRING | Optional | Comma separated list of columns in the phenotype file to include in the analysis that contain the event times |
--phenoExcludeList |
STRING | Optional | Comma separated list of phenotypes to ignore from the analysis |
--covarFile |
FILE | Optional | Covariates file |
--covarCol |
STRING | Optional | Use for each covariate you want to include in the analysis |
--covarColList |
STRING | Optional | Comma separated list of covariates to include in the analysis |
--catCovarList |
STRING | Optional | Comma separated list of categorical covariates to include in the analysis |
--covarExcludeList |
STRING | Optional | Comma separated list of covariates to ignore |
--pred |
FILE | Optional | File containing predictions from Step 1 (see Overview). This is required for --step 2 |
--tpheno-file |
STRING | Optional | to use a phenotype file in transposed format (e.g. BED format) |
--tpheno-indexCol |
INT | Optional | index of phenotype name column in transposed phenotype file |
--tpheno-ignoreCols |
INT | Optional | indexes of columns to ignore in transposed phenotype file |
--iid-only |
FLAG | Optional | to specify if header in transposed phenotype file only contains sample IID (assume FID=IID) |
Note: Parameter expansion can be used when specifying phenotypes/covariates (e.g. --covarCol PC{1:10}).
Also, multiple files can be specified for --extract/--exclude/--keep/--remove by using a comma-separated list.
Genetic data file format
regenie can read BGEN files, bed/bim/fam files or pgen/psam/pvar files in Step 1 and Step 2.
The BGEN file format is described here.
The bed/bim/fam file format is described here.
The pgen/pvar/psam file format is described here.
Tools useful for genetic data file format conversion are : PLINK, QCTOOL, BCFTOOLS.
Step 2 of regenie can be sped up by using BGEN files using v1.2 format with 8 bits encoding
(genotype file can be generated with PLINK2 using
option --export bgen-1.2 'bits=8') as well as having an accompanying .bgi index file
(a useful tool to create such file is bgenix which is part of the BGEN library).
To include X chromosome genotypes in step 1 and/or step 2, males should be coded as diploid so that their genotypes are 0/2 (this is done automatically for BED and PGEN file formats with haploid genotypes). Chromosome values of 23 (for human analyses), X, Y, XY, PAR1 and PAR2 are all acceptable and will be collapsed into a single chromosome.
Sample inclusion/exclusion file format
2 2
7 7
.
No header. Each line starts with individual FID IID. Space/tab separated.
Samples listed in the file that are not in bgen/bed/pgen file are ignored.
Variant inclusion/exclusion file format
20
31
.
No header. Each line must start with variant ID (if there are additional columns, file must be space/tab separated).
Variants listed in this file that are not in bgen/bed/pgen file are ignored.
Covariate file format
FID IID V1 V2 V3
1 1 1.46837294454993 1.93779743016325 0.152887004505393
2 2 -1.2234390803815 -1.63408619199948 -0.190201446835255
3 3 0.0711531925667286 0.0863906292357564 0.14254739715665
.
Line 1 : Header with FID, IID and covariate names.
Followed by lines of values. Space/tab separated.
Each line contains individual FID and IID followed by covariate values.
Samples listed in this file that are not in bgen/bed/pgen file are ignored. Genotyped samples that are not in this file are removed from the analysis as well as samples with missing values at any of the covariates included.
If --step 2 is specified, then the covariate file should be the same
as that used in Step 1.
Phenotype file format
FID IID Y1 Y2
1 1 1.64818554321186 2.2765234736685
2 2 -2.67352013711554 -1.53680421614647
3 3 0.217542851471485 0.437289912695016
.
Line 1 : Header with FID, IID and phenotypes names.
Followed by lines of values. Space/tab separated.
Each line contains individual FID and IID followed by P phenotype values
(for binary traits, must be coded as 0=control, 1=case, NA=missing unless using --1).
Samples listed in this file that are not in bgen/bed/pgen file are ignored. Genotyped samples that are not in this file are removed from the analysis.
Missing values must be coded as NA.
With QTs, missing values are mean-imputed in Step 1 and they are dropped when testing each phenotype in Step 2 (unless using --force-impute).
With BTs, missing values are mean-imputed in Step 1 when fitting the level 0 linear ridge regression and they are dropped when fitting the level 1 logistic ridge regression for each trait . In Step 2, missing values are dropped when testing each trait.
To remove all samples that have missing values at any of the phenotypes, use option --strict in Step 1 and 2.
If using the transposed phenotype file format with option --tpheno-file,
the header line must contain subject IDs as "FID_IID",
otherwise use option --iid-only and only include IIDs (so will assume FID=IID).
Predictions file format
Running --step 1 --out foo will produce
- A set of files containing genomic predictions for each phenotype from Step 1 (see Output section below).
- A file called
foo_pred.listlisting the locations of the prediction files.
The file list is needed as an input file when using --step 2
via the --pred option.
It has one line per phenotype (in any order) that specifies the name of the phenotype and its
corresponding prediction file name.
Each phenotype must have exactly one prediction file and phenotype names
must match with those in the phenotype file.
Phenotypes in this file not included in the analysis are ignored.
Each prediction file contains the genetic predictions for the phenotype (space separated).
Line 1 starts with 'FID_IID' followed by $N$ sample identifiers. It is followed by 23 lines containing the genetic predictions for each chromosome (sex chromosomes are collapsed into chromosome 23).
More specifically, each line has $N+1$ values which are the chromosome number followed by the $N$ leave-one chromosome out (LOCO) predictions for each individual.
Samples in this file not in the bed/pgen/bgen input file are ignored. Genotyped samples not present in this file will be ignored in the analysis of the corresponding trait.
Samples with missing LOCO predictions must have their corresponding phenotype value set to missing.
Options
| Option | Argument | Type | Description |
|---|---|---|---|
--step |
INT | Required | specify step for the regenie run (see Overview) [argument can be 1 or 2] |
--qt |
FLAG | Optional | specify that traits are quantitative (this is the default so can be ommitted) |
--bt |
FLAG | Optional | specify that traits are binary with 0=control,1=case,NA=missing |
--t2e |
FLAG | Optional | specify that traits are time-to-event data with 0=censoring,1=event,NA=missing in event column |
-1,--cc12 |
FLAG | Optional | specify to use 1/2/NA encoding for binary traits (1=control,2=case,NA=missing) |
--bsize |
INT | Required | size of the genotype blocks |
--cv |
INT | Optional | number of cross validation (CV) folds [default is 5] |
--loocv |
FLAG | Optional | flag to use leave-one out cross validation |
--lowmem |
FLAG | Optional | flag to reduce memory usage by writing level 0 predictions to disk (details below). This is very useful if the number of traits is large (e.g. greater than 10) |
--lowmem-prefix |
FILE PREFIX | Optional | prefix where to temporarily write the level 0 predictions |
--split-l0 |
PREFIX,N | Optional | split level 0 across N jobs and set prefix of output files of level 0 predictions |
--run-l0 |
FILE,K | Optional | run level 0 for job K in {1..N} specifying the master file created from '--split-l0' |
--run-l1 |
FILE | Optional | run level 1 specifying the master file from '--split-l0' |
--l1-phenoList |
STRING | Optional | to specify a subset of phenotypes to analyze when using --run-l1 |
--keep-l0 |
FLAG | Optional | avoid deleting the level 0 predictions written on disk after fitting the level 1 models |
--print-prs |
FLAG | Optional | flag to print whole genome predictions (i.e. PRS) without using LOCO scheme |
--force-step1 |
FLAG | Optional | flag to run step 1 when >1M variants are used (not recommened) |
--minCaseCount |
INT | Optional | flag to ignore BTs with low case counts [default is 10] |
--apply-rint |
FLAG | Optional | to apply Rank Inverse Normal Transformation (RINT) to quantitative phenotypes (use in both Step 1 & 2) |
--nb |
INT | Optional | number of blocks (determined from block size if not provided) |
--strict |
FLAG | Optional | flag to removing samples with missing data at any of the phenotypes |
--ignore-pred |
FLAG | Optional | skip reading the file specified by --pred (corresponds to simple linear/logistic regression) |
--htp |
STRING | Optional | to output the summary statistics file in the HTP format (string should correspond to cohort name, e.g. 'UKB_450_EUR') |
--exact-p |
FLAG | Optional | avoid capping p-values at 2.2E-307 in the HTP format summary statistics output |
--use-relative-path |
FLAG | Optional | to use relative paths instead of absolute ones for the step 1 output pred.list file |
--use-prs |
FLAG | Optional | flag to use whole genome PRS in --pred (this is output in step 1 when using --print-prs) |
--gz |
FLAG | Optional | flag to output files in compressed gzip format (LOCO prediction files in step 1 and association results files in step 2) [this only works when compiling with Boost Iostream library (see Install tab)]. |
--force-impute |
FLAG | Optional | flag to keep and impute missing observations for QTs in step 2 |
--write-samples |
FLAG | Optional | flag to write sample IDs for those kept in the analysis for each trait in step 2 |
--print-pheno |
FLAG | Optional | flag to write phenotype name in the first line of the sample ID files when using --write-samples |
--firth |
FLAG | Optional | specify to use Firth likelihood ratio test (LRT) as fallback for p-values less than threshold |
--approx |
FLAG | Optional | flag to use approximate Firth LRT for computational speedup (only works when option --firth is used) |
--firth-se |
FLAG | Optional | flag to compute SE based on effect size and LRT p-value when using Firth correction (instead of based on Hessian of unpenalized log-likelihood) |
--write-null-firth |
FLAG | Optional | to write the null estimates for approximate Firth [can be used in step 1 or 2] |
--compute-all |
FLAG | Optional | to write the null Firth estimates for all chromosomes (regardless of the genotype file) |
--use-null-firth |
FILE | Optional | to use stored null estimates for approximate Firth in step 2 |
--spa |
FLAG | Optional | specify to use Saddlepoint approximation as fallback for p-values less than threshold |
--pThresh |
FLOAT | Optional | P-value threshold below which to apply Firth/SPA correction [default is 0.05] |
--test |
STRING | Optional | specify to carry out dominant or recessive test [default is additive; argument can be dominant or recessive] |
--chr |
INT | Optional | specify which chromosomes to test in step 2 (use for each chromosome to include) |
--chrList |
STRING | Optional | Comma separated list of chromosomes to test in step 2 |
--range |
STRING | Optional | specify chromosome region for variants to test in step 2 [format=CHR:MINPOS-MAXPOS] |
--minMAC |
FLOAT | Optional | flag to specify the minimum minor allele count (MAC) when testing variants [default is 5]. Variants with lower MAC are ignored. |
--minINFO |
FLOAT | Optional | flag to specify the minimum imputation info score (IMPUTE/MACH R^2) when testing variants. Variants with lower info score are ignored. |
--sex-specific |
STRING | Optional | to perform sex-specific analyses [either 'male'/'female'] |
--af-cc |
FLAG | Optional | to output A1FREQ in case/controls separately in the step 2 result file |
--no-split |
FLAG | Optional | flag to have summary statistics for all traits output in the same file |
--starting-block |
INT | Optional | to start step 2 at a specific block/set number (useful if program crashes during a job) |
--nauto |
INT | Optional | number of autosomal chromosomes (for non-human studies) [default is 22] |
--maxCatLevels |
INT | Optional | maximum number of levels for categorical covariates (for non-human studies) [default is 10] |
--niter |
INT | Optional | maximum number of iterations for logistic regression [default is 30] |
--maxstep-null |
INT | Optional | maximum step size for logistic model with Firth penalty under the null [default is 25] |
--maxiter-null |
INT | Optional | maximum number of iterations for logistic model with Firth penalty under the null [default is 1000] |
--par-region |
STRING | Optional | specify build code to determine bounds for PAR1/PAR2 regions (can be 'b36/b37/b38/hg18/hg19/hg38' or 'start,end' bp bounds of non-PAR region) [default is hg38] |
--force-qt |
FLAG | Optional | force QT run for binary traits |
--threads |
INT | Optional | number of computational threads to use [default=all-1] |
--debug |
FLAG | Optional | debug flag (for use by developers) |
--verbose |
FLAG | Optional | verbose screen output |
--version |
FLAG | Optional | print version number and exit |
--help |
FLAG | Optional | Prints usage and options list to screen |
When step 1 of regenie is run in low memory mode (i.e. using --lowmem),
temporary files are created on disk (using --lowmem-prefix tmp_prefix determines
where the files are written [as in tmp_prefix_l0_Y1,...,tmp_prefix_l0_YP
for P phenotypes]). If the prefix is not specified, the default is to use the
prefix specified by --out (see below).
These are automatically deleted at the end of the program (unless the run
was not successful in which case the user would need to delete the files)
See the Wiki page for more details on how to run the level 0 models for Step 1 of regenie in parallel.
Output
| Option | Argument | Type | Description |
|---|---|---|---|
--out |
FILE PREFIX | Required | Output files that depends on --step |
A log file file.log of the output is generated.
Using --step 1 --out file
For the phenotypes, files file_1.loco,...,file_P.loco are output with the
per-chromosome LOCO predictions as rows of the files
(following the order of the phenotypes in the phenotype file header).
If option --gz was used, the files will be compressed in gzip format and have extension .loco.gz.
Genotyped individuals specified using option --remove are excluded from this file.
Individuals with missing phenotype values kept in the analysis
are included in the file and have their predictions set to missing.
The list of blup files needed for step 2 (association testing) is written to file_pred.list.
If using --print-prs, files file_1.prs,...,file_P.prs will be written with the
whole genome predictions (i.e. PRS) without using LOCO scheme (similar format as the .loco files).
The list of these files is written to file_prs.list and can be used in step 2 with --pred and
specifying flag --use-prs. Note that as these are not obtained using a LOCO scheme,
association tests could suffer from proximal contamination.
If using option --write-null-firth, the estimates for approximate Firth under the null will be written to files
file_1.firth,...,file_P.firth and the list of these files is written to file_firth.list. This can be
used in step 2 as --use-null-firth file_firth.list. Note that it assumes the same set of covariates are
used in Step 1 and 2.
Using--step 2 --out file
By default, results are written in separate files for
each phenotype
file_<phenotype1_name>.regenie,...,file_<phenotypeP_name>.regenie.
Each file has one line per
SNP along with a header line.
If option --gz was used, the files will be compressed in gzip format and have extension .regenie.gz.
The entries of each row specify chromosome, position, ID, reference allele (allele 0), alternative allele (allele 1), frequency of the alternative allele, sample size and the test performed (additive/dominant/recessive). With BGEN/PGEN files with dosages, the imputation INFO score is provided (IMPUTE info score for BGEN and Mach Rsq for PGEN). Allele frequency, sample size and INFO score, if applicable, are computed using only non-missing samples for each phenotype.
These are followed by the estimated effect sizes (for allele 1 on the original scale), standard errors, chi-square test statistics and p-value. An additional column is included to specify if Firth/SPA corrections failed.
With option --no-split, the summary statistics for all traits are written to a single file file.regenie,
with the same format as above. Additionaly, an accompanying file with the trait names corresponding to Y1,Y2,...
will be generated in ‘file.regenie.Ydict’. Note that allele frequency, sample size and INFO score are computed using
all analyzed samples.
With option --htp, the summary statistics file will follow the HTP format.
If option --write-samples was used, IDs of samples used for each trait will be written in files
file_<phenotype1_name>.regenie.ids,...,file_<phenotypeP_name>.regenie.ids (tab separated, no header).
When using --par-region, the default boundaries used for the chrX PAR regions are:
- b36/hg18: 2709520 and 154584238
- b37/hg19: 2699520 and 154931044
- b38/hg38: 2781479 and 155701383
Gene-based testing
Starting from version 3.0, Step 2 of regenie provides a complimentary set of gene-based test in addition to the burden testing functionality introduced in version 2.0. More specifically, for a given set of variants (eg within a gene) which can be defined using functional annotations, regenie can apply various set-based tests on the variants as well as collapse them into a single combined 'mask' genotype that can be tested for association just like a single variant.
Input
| Option | Argument | Type | Description |
|---|---|---|---|
--anno-file |
FILE | Required | File with variant annotations for each set |
--set-list |
FILE | Required | File listing variant sets |
--extract-sets |
FILE | Optional | Inclusion file that lists IDs of variant sets to keep |
--exclude-sets |
FILE | Optional | Exclusion file that lists IDs of variant sets to remove |
--extract-setlist |
STRING | Optional | Comma-separated list of variant sets to keep |
--exclude-setlist |
STRING | Optional | Comma-separated list of variant sets to remove |
--aaf-file |
FILE | Optional | File with variant AAF to use when building masks (instead of AAF estimated from sample) |
--mask-def |
FILE | Required | File with mask definitions using the annotations defined in --anno-file |
Note: multiple files can be specified for --extract-sets/--exclude-sets by using a comma-separated list.
Annotation input files
The following files are used to define variant sets and functional annotations which will be used to generate masks.
Annotation file
1:55039839:T:C PCSK9 LoF
1:55039842:G:A PCSK9 missense
.
This file defines functional annotations for variants. It is designed to accommodate for variants with separate annotations for different sets/genes.
Each line contains the variant name, the set/gene name and a single annotation category (space/tab separated).
Variants not in this file will be assigned to a default "NULL" category. A maximum of 63 annotation categories (+NULL category) is allowed.
For gene sets, tools you can use to obtain variant annotations per transcripts are snpEFF or VEP. To obtain a single annotation per gene, you could choose the most deleterious functional annotation across the gene transcripts or alternatively use the canonical transcript (note that its definition can vary across software).
We have implemented an extended 4-column format of the annotation file which also categorizes sets into domains (e.g. for gene sets, these would correspond to gene domains).
1:55039839:T:C PCSK9 Prodomain LoF
1:55039842:G:A PCSK9 Prodomain missense
.
Masks will be generated for each domain (maximum of 8 per set/gene) in addition to a mask combining across all domains. Variants can only be assigned to a single domain for each set/gene.
Starting with v4.1, you can also specify custom variant weights which will be used in the burden, SKAT/SKAT-O and ACAT-V tests ($w_i$'s in the gene-based testing overview). Multiple weights can be included in the annotation file after the 3rd column, e.g.
1:55039839:T:C PCSK9 LoF 0.9 0.812 1
1:55039842:G:A PCSK9 missense 0.4 0.23 0.55
.
Using --weights-col 4 will use weights in the 4-th column for the gene-based tests.
Set list file
This file lists variants within each set/gene to use when building masks. Each line contains the set/gene name followed by a chromosome and physical position for the set/gene, then by a comma-separated list of variants included in the set/gene.
A1BG 19 58346922 19:58346922:C:A,19:58346924:G:A,...
A1CF 10 50806630 10:50806630:A:G,10:50806630:A:AT,...
.
Set inclusion/exclusion file format
The file must have a single column of set/gene names corresponding to those in the set list file.
PIGP
ZBTB38
.
AAF file (optional)
Both functional annotations and alternative allele frequency (AAF) cutoffs are used when building masks (e.g. only considering LoF sites where AAF is below 1%). By default, the AAF for each variant is computed from the sample but alternatively, the user can specify variant AAFs using this file.
Each line contains the variant name followed by its AAF (it should be for the ALT allele used in the genetic data input). AAF must be a numerical value (i.e. it cannot be '.').
7:6187101:C:T 1.53918207864341e-05
7:6190395:C:A 2.19920388819247e-06
.
Since singleton variants cannot be identified from this file, they are determined by default
based on the input genetic data. To enforce which sites should be included in the singleton masks
(see --set-singletons), you can add a third column in the file with a binary indicator
(1=singleton; 0=not singleton). So only variants which are specified as singletons will be
considered for the singleton masks, regardless of whether they are singletons in the input genetic data.
Note that with this flag, singleton sites will be included in all masks (regardless of the AAF in file).
7:6187101:C:T 1.53918207864341e-05 0
7:6190395:C:A 2.19920388819247e-06 1
.
Mask definitions
Mask file
This file specifies which annotation categories should be combined into masks. Each line contains a mask name followed by a comma-seperated list of categories included in the mask (i.e. union is taken over categories).
For example below, Mask1 uses only LoF variants and Mask2 uses LoF and missense annotated variants.
Mask1 LoF
Mask2 LoF,missense
.
AAF cutoffs
Option --aaf-bins specifies the AAF upper bounds used to generate burden masks
(AAF and not MAF [minor allele frequency] is used when deciding which variants go into a mask).
By default, a mask based on singleton sites are always included.
For example, --aaf-bins 0.01,0.05 will generate 3 burden masks for AAFs in
[0,0.01], [0,0.05] and singletons.
SKAT/ACAT tests
The option --vc-tests is used to specify the gene-based tests to run.
By default, these tests use all variants in each mask category.
If you'd like to only include variants whose AAF is below a given threshold
,e.g. only including rare variants, you can use --vc-maxAAF.
| Test | Name in regenie | Description |
|---|---|---|
| SKAT | skat | Variance component test |
| SKATO | skato | Omnibus test combining features of SKAT and Burden |
| SKATO-ACAT | skato-acat | Same as SKATO but using Cauchy combination method to maximize power across SKATO models |
| ACATV | acatv | Test using Cauchy combination method to combine single-variant p-values |
| ACATO | acato | Omnibus test combining features of ACATV, SKAT and Burden |
| ACATO-FULL | acato-full | Same as ACATO but using the larger set of SKATO models used in the SKATO test |
For example, --vc-tests skato,acato-full will run SKATO and ACATO
(both using the default grid of 8 rho values for the SKATO models) and
the p-values for SKAT, SKATO, ACATV and ACATO will be output.
Ultra-rare variants (defined by default as MAC$\le$10, see --vc-MACthr) are collapsed into
a burden mask which is then included in the tests instead of the individual variants.
For additional details on the tests, see here.
Joint test for burden masks
The following tests can be used to combine different burden masks generated using different annotation classes as well as AAF thresholds.
| Test | Name in regenie | QT | BT | Robust to LD | Assumes same effect direction |
|---|---|---|---|---|---|
| Minimum P-value | minp | $\checkmark$ | $\checkmark$ | $\times$ | $\times$ |
| ACAT | acat | $\checkmark$ | $\checkmark$ | $\checkmark$ | $\times$ |
| SBAT | sbat | $\checkmark$ | $\times$ | $\checkmark$ | $\checkmark$ |
The ACAT test combines the p-values of the individual burden masks using the Cauchy combination method (see ref. 14 here). The SBAT test is described into more detail here.
If you only want to output the results for the joint tests (ignore the marginal tests), use --joint-only.
LOVO/LODO schemes
The leave-one-variant-out (LOVO) scheme takes all sites going into a mask, and builds LOVO masks by leaving out one variant at a time from the full set of sites. The mask including all sites will also be computed.
The argument for --mask-lovo is a comma-separated list which
consists of
the set/gene name,
the mask name,
and the AAF cutoff (either 'singleton' or a double in (0,1)).
If using a 4-column annotation file, then --mask-lovo should have
the gene name,
the domain name,
the mask name,
and the AAF cutoff.
So the LOVO masks will be generated for a specific gene domain.
The leave-one-domain-out (LODO) scheme (specified by --mask-lodo)
takes all sites going into a mask and builds a LODO mask for each domain specified for the gene
by excluding all variants in the domain.
The full mask including all sites will also be computed.
The argument for --mask-lodo should have the gene name, the mask name and the AAF cutoff.
Writing mask files
Burden masks built in regenie can be written to PLINK bed format. If the input genetic data contains dosages, the masks dosages will be converted to hard-calls prior to being written to file and these hard-calls will be used for the association testing.
The PLINK bed file is written using 'ref-last' encoding (i.e. REF allele is listed last in the bim file).
Note that this cannot be used with the LOVO/LODO schemes.
Options
| Option | Argument | Type | Description |
|---|---|---|---|
--aaf-bins |
FLOAT,...,FLOAT | Optional | comma-separated list of AAF upper bounds to use when building masks [default is a single cutoff of 1%] |
--build-mask |
STRING | Optional | build masks using the maximum number of ALT alleles across sites ('max'; the default), or the sum of ALT alleles ('sum'), or thresholding the sum to 2 ('comphet') |
--singleton-carrier |
FLAG | Optional | to define singletons as variants with a single carrier in the sample (rather than alternative allele count=1) |
--set-singletons |
FLAG | Optional | to use 3rd column in AAF file to specify variants included in singleton masks |
--write-mask |
FLAG | Optional | write mask to PLINK bed format (does not work when building masks with 'sum') |
--vc-tests |
STRING | Optional | comma-separated list of SKAT/ACAT-type tests to run |
--vc-maxAAF |
FLOAT | Optional | AAF upper bound to use for SKAT/ACAT-type tests [default is 100%] |
--skat-params |
FLOAT,FLAT | Optional | a1,a2 values for the single variant weights computed from Beta(MAF,a1,a2) used in SKAT/ACAT-type tests [default is (1,25)] |
--skato-rho |
FLOAT,...,FLOAT | Optional | comma-separated list of $\rho$ values used for SKATO models |
--vc-MACthr |
FLOAT | Optional | MAC threshold below which to collapse variants in SKAT/ACAT-type tests [default is 10] |
--joint |
STRING | Optional | comma-separated list of joint tests to apply on the generated burden masks |
--rgc-gene-p |
FLAG | Optional | to compute the GENE_P test |
--skip-test |
FLAG | Optional | to skip computing association tests after building masks and writing them to file |
--mask-lovo |
STRING | Optional | to perform LOVO scheme |
--lovo-snplist |
FILE | Optional | File with list of variants for which to compute LOVO masks |
--mask-lodo |
FLAG | Optional | to perform LODO scheme |
--weights-col |
INT | Optional | column index (1-based) in annotation file to use custom weights in gene-based tests |
--write-mask-snplist |
FLAG | Optional | to write list of variants that went into each mask to file |
--check-burden-files |
FLAG | Optional | to check the concordance between annotation, set list and mask files [see below] |
--strict-check-burden |
FLAG | Optional | to exit early if the annotation, set list and mask definition files dont agree [see below] |
Three rules can be used to build masks with --build-mask as shown in diagram below,
where the last rule comphet applies a threshold of 2 to the mask from the sum rule.

Output
With --out file
Results are written in separate files for each phenotype
file_<phenotype1_name>.regenie,...,file_<phenotypeP_name>.regenie
with the same output format mentioned above.
Additionally, a header line is included (starting with ##)
which contains mask definition information.
Masks will have name <set_name>.<mask_name>.<AAF_cutoff> with the
chromosome and physical position having been defined in the set list file,
and the reference allele being ref, and the alternate allele corresponding to
<mask_name>.<AAF_cutoff>.
When using --mask-lovo, the mask name will be the same as above but have suffix
_<variant_name> to specify the variant which was excluded when building the mask.
With --build-mask sum, the reported mask AAF corresponds to the average
AAF across sites included in the mask.
If using --write-mask, the masks will be saved to
file_masks.{bed,bim,fam} and if using --write-mask-snplist,
the list of variants included in each mask will be saved to file_masks.snplist.
When using --rgc-gene-p, it will apply the single p-value per gene GENE_P strategy
using all masks (see here for details).
Example run
Using Step 1 results from the Step 1 command above, we use the following command to build and test masks in Step 2
./regenie \
--step 2 \
--bed example/example_3chr \
--covarFile example/covariates.txt \
--phenoFile example/phenotype_bin.txt \
--bt \
--remove example/fid_iid_to_remove.txt \
--firth --approx \
--pred fit_bin_out_pred.list \
--anno-file example/example_3chr.annotations \
--set-list example/example_3chr.setlist \
--mask-def example/example_3chr.masks \
--aaf-bins 0.1,0.05 \
--write-mask \
--bsize 200 \
--out test_bin_out_firth
For each set, this will produce masks using 3 AAF cutoffs (singletons, 5% and 10% AAF).
The masks are written to PLINK bed file (in test_bin_out_firth_masks.{bed,bim,fam})
and tested for association with each binary trait using Firth approximate test
(summary stats in test_bin_out_firth_<phenotype_name>.regenie).
Note that the test uses the whole genome regression LOCO PRS from Step 1 of regenie (specified by --pred).
Checking input files
To assess the concordance between the input files for building masks, you can use --check-burden-files which will generate a report in file_masks_report.txt containing:
-
for each set, the list the variants in the set-list file which are unrecognized (not genotyped or not present in annotation file for the set)
-
for each mask, the list of annotations in the mask definition file which are not in the annotation file
Additionally, you can use --strict-check-burden to
enforce full agreement between the three files
(if not, program will terminate) :
-
all genotyped variants in the set list file must be in the annotation file (for the corresponding set)
-
all annotations in the mask definition file must be present in the annotation file
Interaction testing
Starting from regenie v3.0, you can perform scans for interactions (either GxE or GxG).
For GxE tests, the interacting variable should be part of the covariate file
(if it is categorical, specify it in --catCovarList).
For GxG tests, the interacting variant can be part of the input genetic file
or it can be present in an external file (see --interaction-snp-file)
Options
| Option | Argument | Type | Description |
|---|---|---|---|
--interaction |
STRING | Optional | to run GxE test specifying the interacting covariate (see below) |
--interaction-snp |
STRING | Optional | to run GxG test specifying the interacting variant (see below) |
--interaction-file |
FORMAT,FILE | Optional | external genotype file containing the interacting variant [FORMAT can be bed/bgen/pgen and FILE is the file name (bgen) or file prefix (bed/pgen)] |
--interaction-file-sample |
FILE | Optional | accompagnying sample file for BGEN format |
--interaction-file-reffirst |
FLAG | Optional | use the first allele as the reference for BGEN or PLINK BED formats |
--no-condtl |
FLAG | Optional | to print out all the main effects from the interaction model (see Output section below) |
--force-condtl |
FLAG | Optional | to include the interacting SNP as a covariate in the marginal test (see Output section below) |
--rare-mac |
FLOAT | Optional | minor allele count (MAC) threshold below which to use HLM method for QTs [default is 1000] |
For GxE tests where the interacting variable is categorical, you can specify the baseline level using --interaction VARNAME[BASE_LEVEL] (e.g. --interaction BMI[<25]). Otherwise, the first value found in the covariate file will be used as the baseline level.
For GxG tests, the default coding for the interacting variant is additive. If you would like to use dominant/recessive/categorical coding, use --interaction-snp SNP_NAME[dom/rec/cat] (for example with dominant coding, --interaction-snp SNPNAME[dom] will allow for separate effects between carriers vs non-carriers of the interacting variant). The allowed values in the brackets are add/dom/rec/cat.
Output
The result files will contain multiple lines for the same variant corresponding to the different null hypotheses being tested in the interaction model
The suffix in the "TEST" column indicates which hypothesis is being tested:
- "ADD": marginal test where the interacting variable has not been added as a covariate $-$ this corresponds to $H_0: \beta = 0$ given $\alpha=\gamma = 0$
- this is only printed for GxG tests by default, or GxE using
--no-condtl
- this is only printed for GxG tests by default, or GxE using
- "ADD-CONDTL": marginal test where the interacting variable has been added as a covariate (default for GxE tests) $-$ this corresponds to $H_0: \beta = 0$ given $\gamma = 0$
- this is only printed for GxE tests by default, or GxG using
--force-condtl
- this is only printed for GxE tests by default, or GxG using
- "ADD-INT_VAR": test for the main effect of the interaction variable ("VAR" will be replaced by the name of the interacting variable) $-$ this corresponds to $H_0: \alpha = 0$
- this is only printed for GxG tests by default, or GxE using
--no-condtl - If the interacting variable is categorical, you will have separate lines for each level aside from the baseline level (e.g. "ADD-INT_BMI=25-30" and "ADD-INT_BMI=30+" where baseline level is "$<$25")
- will also output the effect of $E^2$ in "ADD-INT_VAR^2" if the trait is binary (see here)
- this is only printed for GxG tests by default, or GxE using
- "ADD-INT_SNP": test for main effect of tested SNP in the interaction model $-$ this corresponds to $H_0: \beta = 0$
- "ADD-INT_SNPxVAR": test for interaction effect ("VAR" will be replaced by the name of the interacting variable) $-$ this corresponds to $H_0: \gamma = 0$
- If the interacting variable is categorical, you will have separate lines for each level aside from the baseline level (e.g. "ADD-INT_SNPxBMI=25-30" and "ADD-INT_SNPxBMI=30+" where baseline level is "$<$25")
- With Firth correction, only the effect sizes for the interaction effect at each level will be reported and the LRT p-value will only be computed for the joint test on the interaction effects
- If the interacting variable is categorical, you will have separate lines for each level aside from the baseline level (e.g. "ADD-INT_SNPxBMI=25-30" and "ADD-INT_SNPxBMI=30+" where baseline level is "$<$25")
- "ADD-INT_$k$DF": joint test for main and interaction effect of tested variant ($k\ge2$ for categorical interacting variables) $-$ this corresponds to $H_0: \beta = \gamma = 0$
Conditional analyses
Starting from regenie v3.0, you can specify genetic variants to add to the set of covariates when performing association testing. This works in both step 1 and 2, and can be used in conjunction with the gene-based tests or the interactiong testing feature. The conditioning variants will automatically be ignored from the analysis.
| Option | Argument | Type | Description |
|---|---|---|---|
--condition-list |
FILE | Required | file with list of variants to condition on |
--condition-file |
FORMAT,FILE | Optional | get conditioning variants from external file (same argument format as --interaction-file) |
--condition-file-sample |
FILE | Optional | accompagnying sample file for BGEN format |
--max-condition-vars |
INT | Optional | maximum number of conditioning variants [default is 10,000] |
Survival analyses
Starting from regenie v4.0, you can conduct survival analysis for time-to-event data.
Phenotype file format
In this small example, there are 5 samples, and the event of interest is the diagnosis of cancer over a period of 10 years.
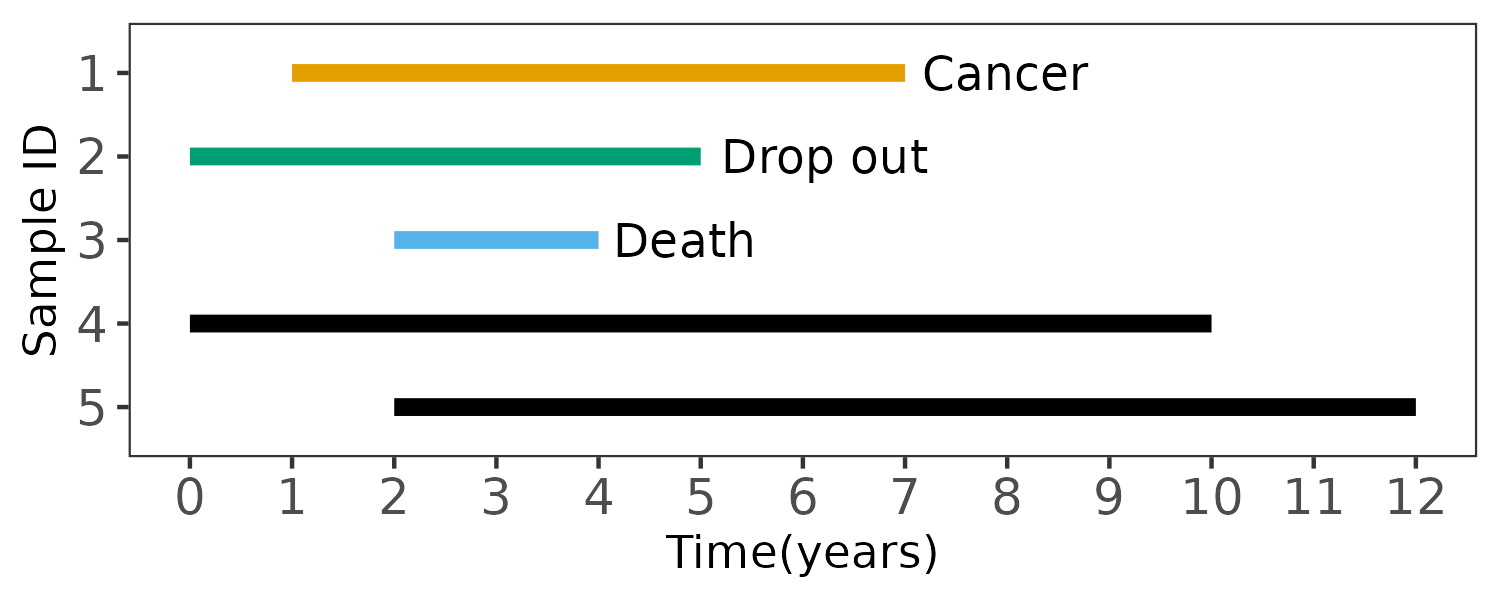
Sample 1 is diagnosed with cancer during the study; the time variable is the number of years until the sample is diagnosed with cancer. Sample 2 drops out of the study; sample 3 dies during the study; sample 4 and 5 complete the study without being diagnosed with cancer; they are all right-censored, and the time variable is the last encounter or death time. The corresponding phenotype file is
FID IID Time Cancer
1 1 6 1
2 2 5 0
3 3 2 0
4 4 10 0
5 5 10 0
Required options
Survival analysis in regenie requires the following specific options in step 1, step 2 and gene-based burden tests.
| Option | Argument | Type | Description |
|---|---|---|---|
--t2e |
FLAG | Required | specify the traits are time-to-event data |
--phenoColList |
STRING | Required | Comma separated list of time names to include in the analysis |
--eventColList |
STRING | Required | Comma separated list of columns in the phenotype file to include in the analysis that contain the events. These event columns should have 0=no event,1=event,NA=missing |
For the example above, the regenie call is
./regenie \
--t2e \
--phenoColList Time \
--eventColList Cancer \
...
For a phenotype file containing multiple time-to-event traits, the order of censor variables listed in --eventColList should match the order of time names specified in --phenoColList. For example, the phenotype file is
FID IID Cancer_Time Cancer Asthma_Time Asthma
1 1 6 1 4 0
2 2 5 0 8 1
The regenie call is
./regenie \
--t2e \
--phenoColList Cancer_Time,Asthma_Time \
--eventColList Cancer,Asthma \
...
The output format is the same as the output file for quantitative and binary traits, with the BETA column containing the estimated harzard ratio (on log scale).
LD computation
REGENIE can calculate LD between a group of variants on the same chromosome.
| Option | Argument | Type | Description |
|---|---|---|---|
--compute-corr |
FLAG | Required | compute LD matrix and write to binary file |
--output-corr-text |
FLAG | Optional | write Pearson correlations to text file |
--forcein-vars |
FLAG | Optional | retain all variants specified in --extract which absent from the genetic data in the LD matrix |
--ld-extract |
FILE | Optional | file listing single variants as well as burden masks to include in LD matrix (see below) |
Note that this can be quite memory intensive for large groups of variants (memory ~$8M^2$ bytes for $M$ variants).
Output
Using--step 2 --out file
By default, the LD matrix is stored in a binary compressed file file.corr and
the list of variants corresponding to the columns of the LD matrix are stored in file.corr.snplist.
The R script scripts/parseLD.r contains a function which returns the LD matrix, e.g. get.corr.sq.matrix("file.corr").
Using --output-corr-text will write the Pearson correlations to a text file instead.
When using --forcein-vars, variants not present in the genetic data will be added as extra column/rows in the LD matrix.
For these variants, the diagonal entries in the matrix will be set to 1 and the off-diagonal entries 0.
Using--ld-extract info.txt
This option is used compute LD between single variants and burden masks generated on-the-fly in REGENIE; it requires specifying annotation files.
The file info.txt should have three columns: variant type ('sv' or 'mask'), variant name, followed by the set (e.g. gene) name (this can be 'NA' for single variant). For example, it would look like:
sv 1:1111:A:G NA
sv 1:2222:C:T NA
mask PCSK9.M1.0.01 PCSK9
.
Note that the set and mask names must match that used in REGENIE based on provided annotation files and allele frequency cutoffs. Variant/masks not present in the data will be kept in the LD matrix but will have the corresponding correlations set to 0.